Creador de Árbol Filogenético Impulsado por IA
Describe los organismos y sus relaciones evolutivas, y nuestra IA generará un árbol filogenético profesional al instante. Ideal para filogenética molecular, taxonomía, epidemiología y biología evolutiva.
Generador de Árbol Filogenético
By using ConceptViz, you agree not to generate or edit adult, sexual, explicit, unsafe, or policy-violating content. See Content Policy.
Gratis para probar ·
Tu árbol filogenético aparecerá aquí
Describe los organismos y las relaciones, luego haz clic en Generar
Ejemplos de Árboles Filogenéticos
Explora ejemplos de árboles filogenéticos de diversos campos o genera el tuyo arriba
Árbol Filogenético de Mamíferos
Filogenia completa de mamíferos que ilustra la radiación de los principales órdenes desde los monotremas hasta los primates, con tiempos de divergencia estimados en millones de años.
Árbol de Evolución de Virus
Filogenia molecular de familias de virus ARN que ilustra las relaciones evolutivas, las tasas de mutación y los eventos de transmisión entre especies.
Filogenia del ARNr 16S Bacteriano
Filogenia del gen ARN ribosomal 16S de los principales filos bacterianos, el marcador molecular estándar utilizado para la clasificación bacteriana y la metagenómica.
Filogenia de la Evolución Vegetal
Árbol evolutivo de plantas terrestres que muestra la progresión desde los ancestros acuáticos a través de briófitas, helechos, gimnospermas, hasta plantas con flores con adaptaciones clave etiquetadas.
Filogenia de la Migración Humana
Filogenia de haplogrupos de ADN mitocondrial que traza los patrones de migración humana desde África hasta todos los continentes habitados, con fechas estimadas de divergencia.
Árbol de Familia de Proteínas
Filogenia molecular de una superfamilia de proteínas que ilustra la divergencia de secuencias, los dominios conservados y la evolución funcional en organismos modelo.
¿Qué es un Árbol Filogenético?
Un árbol filogenético es un diagrama ramificado que representa las relaciones evolutivas entre entidades biológicas — típicamente especies, genes o proteínas. Cada punto de ramificación (nodo interno) representa un ancestro común hipotético, y las puntas (nodos terminales u hojas) representan los taxones que se comparan. Las longitudes de las ramas pueden codificar el tiempo evolutivo (cronogramas), la distancia genética (filogramas), o no tener significado cuantitativo (cladogramas). Los árboles filogenéticos son el marco central de la biología evolutiva, proporcionando una hipótesis visual de cómo los organismos están relacionados a través de la descendencia con modificación. Se construyen utilizando caracteres morfológicos, datos de secuencias moleculares (ADN, ARN o proteínas), o una combinación de ambos.
Tipos de Árboles Filogenéticos
- Árboles con raíz — tienen un solo nodo ancestral (raíz) que representa el ancestro común más reciente de todos los taxones; la dirección de la evolución fluye desde la raíz hasta las puntas
- Árboles sin raíz — muestran las relaciones entre taxones sin especificar el ancestro común ni la dirección de la evolución; comúnmente producidos por métodos de neighbor-joining y máxima verosimilitud
- Cladogramas — las longitudes de las ramas son uniformes y no contienen información; solo la topología (patrón de ramificación) importa para mostrar las características derivadas compartidas
- Filogramas — las longitudes de las ramas son proporcionales a la cantidad de cambio evolutivo (por ej., sustituciones nucleotídicas por sitio)
- Cronogramas — las longitudes de las ramas son proporcionales al tiempo, con las puntas alineadas al presente; calibrados usando registros fósiles o estimaciones de reloj molecular
- Árboles circulares (radiales) — un árbol con raíz dibujado en disposición circular para mostrar eficientemente un gran número de taxones, comúnmente utilizado en metagenómica y genómica comparativa
Cómo Leer un Árbol Filogenético
Leer correctamente un árbol filogenético requiere concentrarse en el patrón de ramificación en lugar del orden de los taxones en las puntas. Dos taxones están más estrechamente relacionados si comparten un ancestro común más reciente (nodo) que excluye a otros taxones. La rotación de las ramas alrededor de un nodo no cambia las relaciones evolutivas — solo importa la topología. Los valores de bootstrap (típicamente 0-100) o las probabilidades posteriores en los nodos indican el soporte estadístico para ese arreglo de ramificación particular. Valores más altos significan mayor confianza en que la agrupación es correcta. Las longitudes de las ramas en los filogramas representan la cantidad de cambio evolutivo; ramas más largas indican mayor divergencia genética. Una barra de escala muestra la unidad de medida, como sustituciones por sitio nucleotídico. Los grupos externos — taxones que se sabe están fuera del grupo de interés — se utilizan para enraizar el árbol y establecer la dirección de la evolución.
Métodos de Filogenética Molecular
- Alineamiento de secuencias — el primer paso, alineando secuencias homólogas de ADN, ARN o proteínas para identificar posiciones conservadas y variables utilizando herramientas como MUSCLE, MAFFT o ClustalW
- Métodos basados en distancias — calculan distancias evolutivas por pares y agrupan los taxones en consecuencia; neighbor-joining (NJ) es rápido y ampliamente utilizado para análisis exploratorio
- Máxima parsimonia — encuentra el árbol que requiere el menor número de cambios evolutivos para explicar los datos de secuencias observados; efectivo para taxones estrechamente relacionados
- Máxima verosimilitud (ML) — evalúa la probabilidad de los datos de secuencias dado una topología de árbol y un modelo de sustitución; estadísticamente riguroso pero computacionalmente intensivo (por ej., RAxML, IQ-TREE)
- Inferencia bayesiana — utiliza muestreo de Monte Carlo por cadenas de Markov (MCMC) para estimar las probabilidades posteriores de las topologías de árboles dados los datos y un modelo previo (por ej., MrBayes, BEAST)
- Análisis de bootstrap — remuestrea columnas de alineamiento con reemplazo para evaluar la robustez de cada nodo; valores superiores al 70-80% se consideran generalmente bien soportados
Aplicaciones en la Investigación Biológica
- Taxonomía y sistemática — clasificación de organismos y revisión de la nomenclatura taxonómica basada en relaciones evolutivas en lugar de similitud superficial
- Epidemiología y salud pública — rastreo del origen y propagación de patógenos, identificación de eventos de transmisión zoonótica y seguimiento de la evolución de variantes virales en tiempo real
- Biología de la conservación — identificación de especies evolutivamente distintas y globalmente en peligro (EDGE) para priorizar esfuerzos de conservación y preservar la máxima diversidad filogenética
- Descubrimiento de fármacos y genómica funcional — predicción de la función de genes y proteínas a través de ortología filogenética, guiando la búsqueda de dianas terapéuticas entre especies
- Biogeografía y paleontología — reconstrucción de rangos geográficos ancestrales e integración de puntos de calibración fósil para datar eventos evolutivos
- Metagenómica e investigación del microbioma — clasificación de comunidades microbianas utilizando filogenias de ARNr 16S/18S y comprensión de la estructura de comunidades ecológicas
Preguntas frecuentes
Más Herramientas de Biología
 Investigación
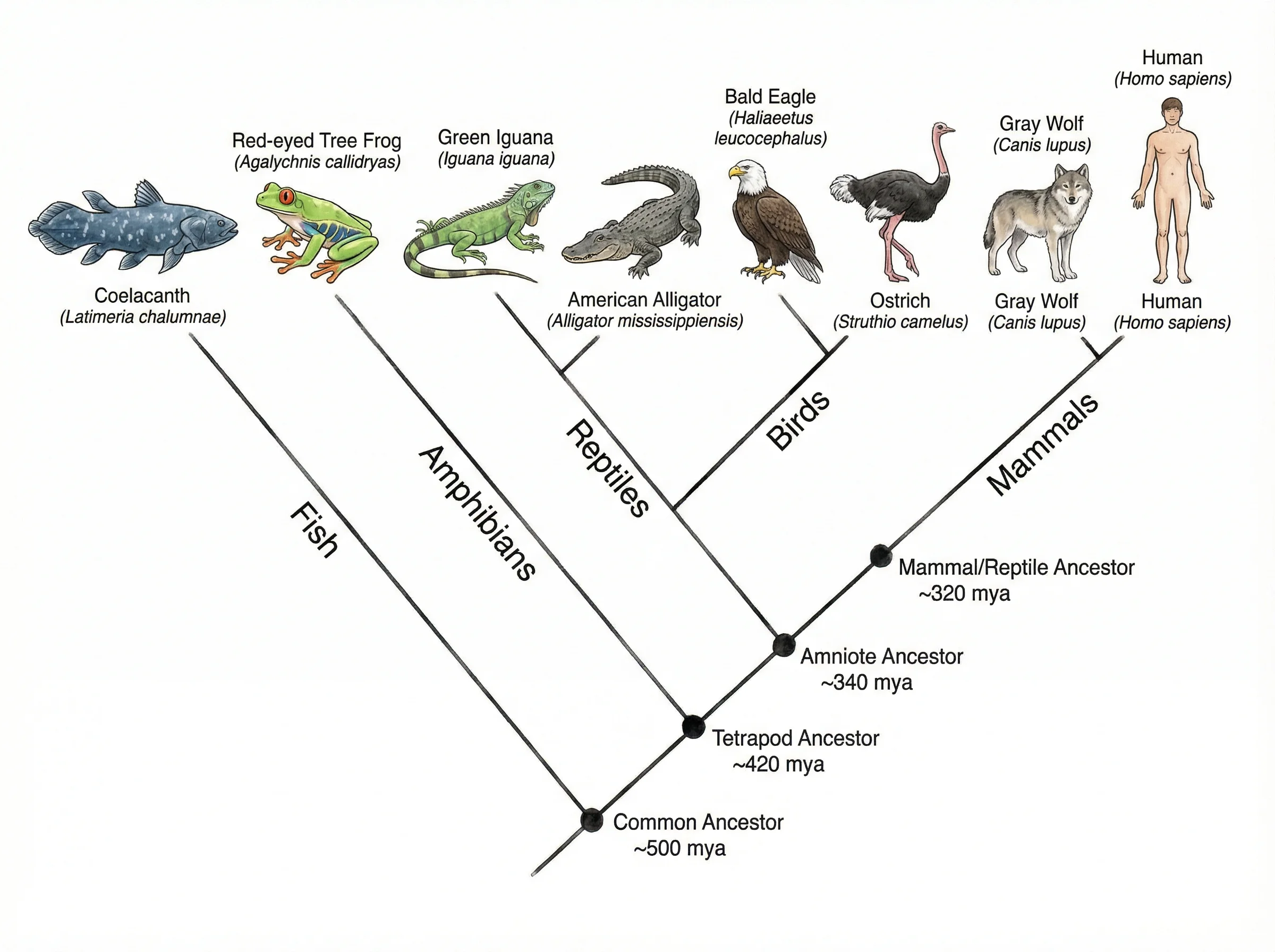
InvestigaciónGenerador de Diagramas de Árbol
Crea diagramas de árbol jerárquicos para taxonomía, clasificación, árboles de decisión y probabilidad.
 Biología
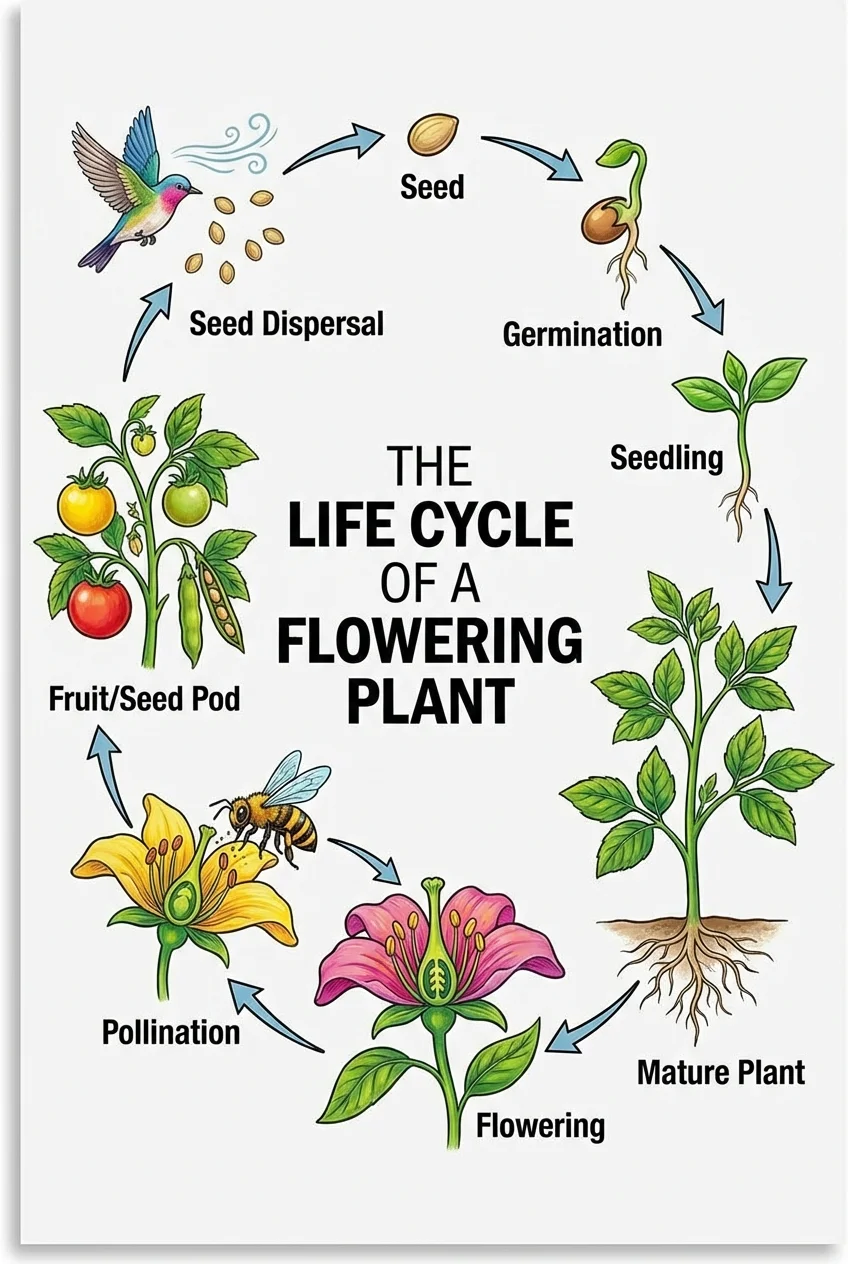
BiologíaGenerador de Diagramas de Ciclo de Vida
Genera diagramas de ciclo de vida para plantas, animales y otros organismos con etapas etiquetadas.
 Investigación
InvestigaciónGenerador de Imágenes Científicas con IA
Crea ilustraciones y diagramas científicos profesionales para artículos de investigación y presentaciones.